Spotters A6

Slide 2
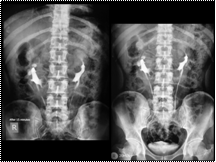
DONKEY SHOE KIDNEY
Slide 3
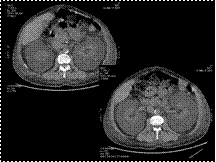
Renal lymphangiectasia
D/D- nephroblastomatosis, polycystic ds, lymphoma, urinoma, abscess
Slide 4
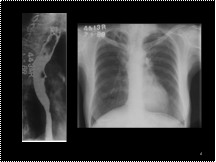
Traction diverticula
Slide 5
Hydrothorax in a fetus at 32 weeks gestation
Slide 6
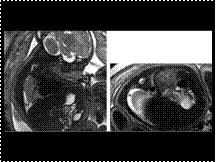
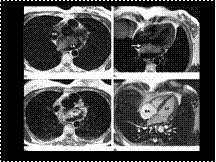
Myxoma of the left atrium
Slide 7
ASCARIASIS
Slide 8
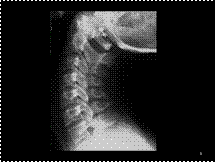
Clay-shoveler's fracture
Slide 9
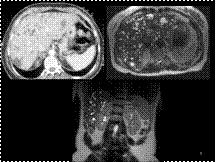
CT demonstrates innumerable, non-enhancing, lesions throughout the liver, with attenuation and signal like that of cysts. Of note there are no cysts in the kidneys or pancreas.
MRI shows innumerable cystic lesions throughout the liver. These range in size from a few millimeters to 2.5 cm in diameter. There appear to be several areas of minimal to mild dilatation of bile ducts throughout the liver. Several of the cyst appear to communicate with adjacent dilated ducts. There is no instrinsic intra or extrahepatic duct dilatation.
Multiple biliary hamartomas
Caroli's disease
Hepatic cysts in association with autosomal-dominant polycystic kidney disease
Metastatic disease to the liver
Hepatic microabscesses
Slide 10
Ulcerative colitis
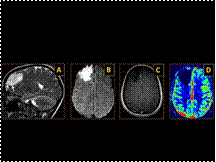
Slide 11
Epidermoid Cyst Epidermoid cysts arise from inclusion of the ectodermal epithelial elements at the time of neural tube closure. Account for up to 1% of all primary intracranial tumors. Commonly seen in young adults. Mostly intradural, cerebellopontine angle cistern is the most common location, followed by the supra and parasellar regions. Key Diagnostic Features: Extra-axial mass predominantly hypointense on T1WI, hyperintense on FLAIR and T2WI, demonstrating restricted diffusion, and no intrinsic enhancement following contrast administration is seen. The mass has a cheesy consistency and therefore insinuates into adjacent cisterns engulfing vessels and nerves. DDx: Arachnoid cyst, infectious/inflammatory cyst such as neurocysticercosis, cystic neoplasm Treatment: Surgical resection
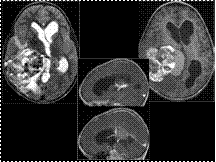
Slide 12
Giant Cavernous Malformation
Prevalence in population of CMs: 0.4%
Giant CMs are rare, may be infiltrative
CMs may be sporadic or associated with previous irradiation, anomalous venous drainage, venous angioma (as in this case).
Syndromes such as familial CMs (defects in chromosomes 7q21.2 & 7p15-p13) or metameric vascular ones
Giant CMs: over 6 cm in diameter (in the literature most have been between 10-13 cm)
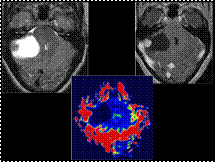
Slide 13
Hemangioblastoma
Most begin as solid masses and then become cystic.
If they remain solid, they tend to be asymptomatic.
If they become cystic, they produce symptoms due to mass effect.
Most common primary cerebellar tumor in adults
35% of patients with cerebellar hemangioblastomas will have von Hippel-Lindau disease.
Sporadic tumors tend to be discovered around 50-60 years of age while syndromic ones tend to be found about 20 years earlier
Slide 14
Klippel-Feil Syndrome
Clinical triad (52%): low hairline, limited neck motion (due to any type of cervical vertebral fusions, 2 or more segments)
Incidence: 1/42,000, females>males, known since Antiquity (Tutankhamen probably had it)
CNS associated lesions: syringomyelia, callosal dysgenesis, meningocele, spinal stenosis, posterior fossa epi-/dermoids
Other associations: Sprengel's scapular deformity ('Angel's' scapulae), omovertebral bones, accessory ribs, anomalies of the great vessels, cleft palate
Slide 15
Myxopapillary Sacrococcygeal Ependymoma
Ependymomas rarely occur outside the CNS. Of those that do, the majority are in the sacrococcygeal or presacral areas.
Primary subcutaneous extraneural sacrococcygeal ependymomas are thought to arise from ependymal rests or from the coccygeal medullary vestige, an ependymal lined remnant at the caudal portion of the neural tube.
Up to 20% develop distant metastasis, which can occur as late as 20 years after original presentation. Therefore, long-term follow-up is necessary.
The subcutaneous sacrococcygeal ependymomas grow slowly and usually present as large masses.
They are generally encapsulated with a firm/rubbery texture.
Key Diagnostic Features: Imaging appearances vary as they are often complicated by hemorrhage, necrosis and may have areas of fibrosis and calcification, which produces a heterogenous appearance on all MR sequences.
DDx: Although appearances mimic those of teratoma, sacrococcygeal teratomas are more common in the newborn. Differential diagnoses in an adult include pilonidal cyst and neurogenic tumors
Slide 16
PAGET'S DISEASE: BASILAR INVAGINATION
Slide 17
Osmotic Demyelination Syndrome
Rapid correction of chronic osmolar imbalance, especially in the presence of concurrent malnutrition, can cause rapid demyelination. This can also occur with normal serum sodium.
This has classically been described as affecting the central pons (central pontine myelinolysis). However, it also commonly affects other white matter areas (extra pontine myelinolysis) and causes demyelination typically in the basal ganglia, corpus callosum and cerebral white matter.
Patients can present with an altered mental state, bradypnea, spastic paraparesis and even a ""locked in"" syndrome.
Treatment is supportive, with survivors likely to require an extended period of neurorehabilitation
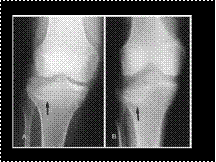
Slide 18
BUMPER OR FENDER FRACTURE
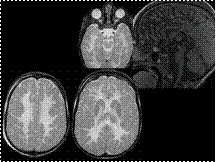
Slide 19
Pelizaeus-Merzbacher Disease (PMD)
Pelizaeus-Merzbacher Disease is a rare leukodystrophy which results from abnormality of PLP1 gene on chromosome Xq22 coding for proteolipid protein 1 and a smaller protein (DM20), two primary components of myelin.
PMD has a ""classic"" form as well a more severe form known as ""connatal"" form. The classic form shows X-linked recessive inheritance, and patients usually present in first year of life with nystagmus, delayed motor and cognitive milestones, and ataxia. The connatal form shows either autosomal or X-linked recessive inheritance and usually presents at birth or in early infancy. In addition to nystagmus and extrapyramidal hyperkinesia, patients have spasticity, optic atrophy and seizure. Patients with classic form usually live until late adolescence or early adulthood, but in connatal form, death usually occurs in early childhood.
There is a group of patients with similar clinical presentation but normal appearing white matter at biopsy and on MRI and no evidence of mutation of PLP1 gene. These group of patients are considered to have Pelizaeus-Merzbacher-like disease.
Key Diagnostic Features: PMD appears as low signal intensity white matter on CT scan, and on MRI, it appears as lack of myelination as opposed to myelin destruction. Depending on the severity of the disease, there might be some myelination within internal capsules, optic radiations and proximal corona radiata. In severe cases, there could be complete absence of low T2 signal intensity within the white matter. The cerebellum may be markedly atrophic. MRI cannot differentiate classic form from connatal form although it has been noted that some degree of myelination is seen in patients with the classic form, whereas no evidence of myelin is seen in connatal form.
MR spectroscopy findings in PMD are controversial likely because of different mutations of the PLP gene. Molecular diagnostic testing is the definitive method to diagnose PMD by detecting mutations of the PLP1 gene.
Treatment: No specific treatment for PMD is available and medical care is mostly limited to supportive care such as physical therapy, anti-spasticity agents, and orthotics.
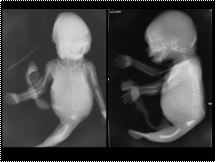
Slide 20
SIRENOMELIA OR MERMAID SYNDROME
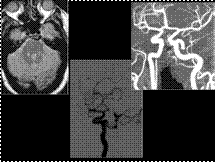
Slide 21
Persistent Trigeminal Artery
Persistent trigeminal artery (PTA) is the most common persistent carotid-basilar arterial anastomosis with an incidence of 0.1 - 0.2%.
PTA is a developmental anomaly in which the short, wide fetal connection between the cavernous carotid and upper third of the basilar artery does not regress.
Other primitive fetal anastamoses include—in consecutive order of appearance—the hypoglossal, proatlantal and otic (acoustic).
The clinical relevance of a PTA is debatable as most cases are discovered incidentally.
Knowledge of the presence of a PTA is vital before performing intracranial sellar-parasellar or vascular intervention.
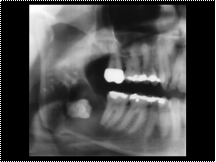
Slide 22
Dentigerous cyst in a 42-year-old man with painful third molars. A full-mouth radiographic series showed an abnormality; additional radiographic views were then obtained. (a) Panoramic radiograph shows an ellipsoid, expansile, well-defined, corticated, lucent lesion with undulating margins in the right mandible. An associated tooth is seen within the lesion. (b) Posteroanterior radiograph shows lingual expansion (arrow). The patient underwent tooth extraction and enucleation. The differential diagnosis includes dentigerous cyst, odontogenic keratocyst, and ameloblastoma.
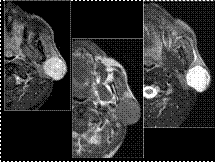
Slide 23
Pleomorphic Adenoma of the Parotid Gland
Most common tumor of parotid gland (50-80%), usually in superficial lobes, may also be found in submandibular and sublingual glands. Most common in adult women (3:2).
MRI: high T2 signal, circumscribed thin low T2 signal fibrous capsule, lobulated contour. May have internal calcifications and cystic degeneration; generally enhance following contrast.
Treated with surgical excision because malignant degeneration can occur.
Differential includes Warthin tumor, monomorphic adenoma, myoepithelioma, oncocytoma,or schwannoma. If cystic, differential includes lymphoepithelial lesions, branchial cleft cyst, simple cyst, or sialocele. In pediatric patients, includes hemangioma or lymphangioma. If bilateral, also consider underlying sarcoid, Sjögren's, or HIV.
Malignant parotid masses typically have intermediate or low T2 signal and an indistinct margin and include mucoepidermoid carcinoma and adenoid cystic carcinoma, metastases, and lymphoma
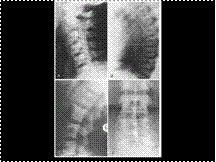
Slide 24
OSTEOPETROSIS: BONE WITHIN A BONE APPEARANCE.
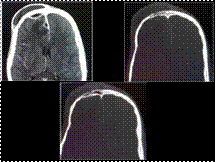
Slide 25
Pott's Puffy Tumor
Pott's puffy tumor is defined as scalp swelling due to subperiosteal abscess resulting from osteomyelitis of the frontal bone secondary to bacterial sinusitis.
Pott first described a puffy indolent tumor of the forehead due to subperiosteal abscess of the frontal bone following trauma.
Undiagnosed or partially treated frontal sinusitis may lead to destruction of outer and inner tables and subperiosteal/intracranial abscess formation. Due to venous communication between frontal bone and dural venous sinuses, intracranial empyema may result without bone destruction. Complications include preseptal and orbital cellulitis resulting from downward spread, intracranial infection in the form of extra-axial epidural and subdural collections, and intraparenchymal abscess formation resulting from posterior extension.
Treatment: excision of the puffy lump, curettage of the osteomyelitic bone, evacuation of intracranial abscess and parentral antibiotics covering aerobes/anaerobes
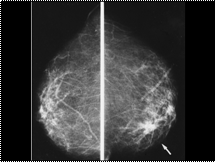
Slide 26
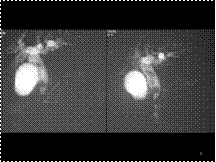
Invasive cancer is frequently more dense than an equal volume of fibroglandular tissue. The invasive cancer in the right breast
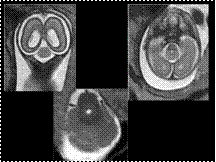
Slide 27
Rhombencephalosynapsis
Rhombencephalosynapsis is a rare congenital malformation characterized by hypogenesis or agenesis of the vermis, fusion of cerebellar hemispheres, dentate nuclei, and superior cerebellar peduncles.
MRI findings include: fused cerebellar hemispheres and dentate nuclei, absent or severely hypoplastic vermis (axial and coronal images), lack of visualization of primary vermian fissure, and lack of a normal fastigial point of the fourth ventricle on sagittal images.
Associated malformations: midline anomalies (aqueductal stenosis, absent septum pellucidum, holoprosencephaly, callosal dysgenesis or agenesis), heterotopias, encephaloceles, clefts, and facial anomalies.
Prenatal identification of rhombencephalosynapsis is useful for counseling since prognosis is poor. Ataxia, involuntary head movements, developmental delay, self injurious behavior, and early death are postnatal presentations.
Slide 28
OSTEOPOIKILOSIS
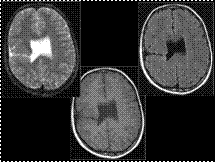
Slide 29
Schizencephaly consists of unilateral or bilateral cleft(s) in the cerebral hemisphere(s) causing communication between the ventricles and subaracnoid spaces.
Clefts are often found in perisilvian areas but can be found all along the cerebral cortex and are lined with gray matter (usually polymicrogyria).
Two variations: the first is open lip where the walls of the cleft are widely separated. The second is closed lip where the walls are in close approximation.
There is often associated septo-optic dysplasia with agenesis of the septum pellucidum and optic nerve hypoplasia.
The ventricle wall may be tented and ""point"" to the defect.
The etiology of schizencephaly is unclear and likely multifactorial.
Clinical findings include focal seizures with early onset and developmental delay and contra-lateral paraplegia
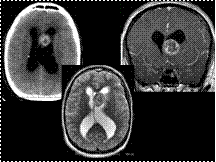
Slide 30
Subependymal Giant Cell Astrocytoma in Tuberous Sclerosis
Subependymal Giant Cell Astrocytoma (SEGA) is the most common cerebral neoplasm in TS, a disease that has an autosomal dominant pattern of inheritance in 20'50% of patients and the presence of tubers and subependymal glial nodules in 90'100%.
SEGA is virtually always located near the foramina of Monro; symptoms relate to increased intracranial pressure and seizures.
SEGA is characterized by slow growth and benign biologic behavior corresponding to WHO grade I.
On CT, SEGA appears as an isoattenuated to slightly hypoattenuated mass near a foramen of Monro. Calcification is common. Hemorrhage within a SEGA may also occur.
On MRI, SEGA usually manifests with T1 hypointensity compared to white matter and heterogeneous hyperintensity on T2 sequences.
Slide 31
MYODIL DYE
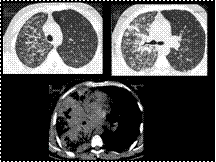
Slide 33
Broncho-alveolar carcinoma
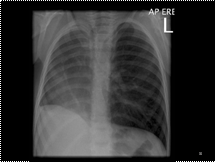
Slide 34
Scimitar syndrome is characterised by a hypoplastic lung that is drained by an anomalous vein into the systemic venous system. It is essentially a combination of pulmonary hypoplasia and partial anomalous pulmonary venous return. It almost exclusively occurs on the right side. The haemodynamics are that of an acyanotic left to right shunt. The anomalous vein most commonly drains into the IVC, right atrium or portal vein. The lung is frequently perfused by the aorta, but the bronchial tree is still connected and thus the lung is not sequestered. CXR findings are that of a small lung with ipsilateral mediastinal shift, and in one third of cases the anomalous draining vein may be seen as a tubular structure paralleling the right heart border in the shape of a Turkish sword (“scimitar”). Scimitar syndrome is associated with congenital heart disease, ipsilateral diaphragmatic anomalies and vertebral anomalies.
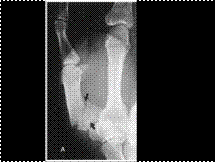
Slide 35
ROLANDO'S FRACTURE
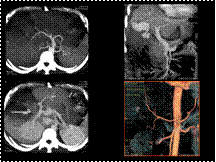
Slide 36
RUPTURED CYSTIC ARTERY
PSEUDOANEURYSM
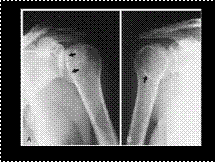
Slide 37
POSTERIOR SHOULDER DISLOCATION
Slide 38
Hydatid cyst peritoneum
Slide 39
GIST
Slide 40
Neuroblastoma
Slide 41
Renal A. aneurysm
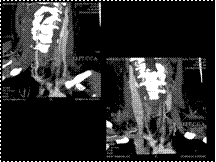
Slide 42
TAKAYASU
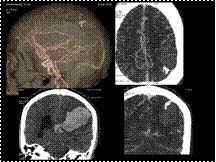
Slide 43
Bleed with AV Malformation
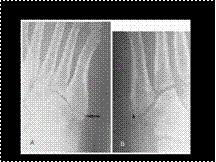
Slide 44
JONES FRACTURE VERSUS NORMAL VARIANT
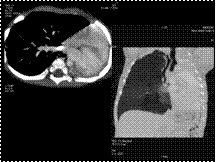
Slide 45
PUL AGENESIS
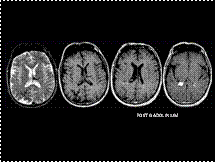
Slide 46
STURGE WEBER
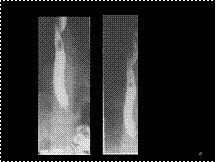
Slide 47
Aberrant R subclavian artery
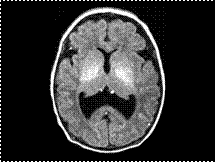
Slide 48
LOBAR HOLOPROSENCEPHALY
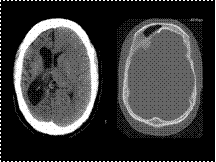
Slide 49
Differential diagnosis of cerebral hemiatrophy
Dyke-Davidoff-Masson Syndrome.
Rasmussen Encephalitis.
Sturge Weber Syndrome.
Hemimegalencephaly.
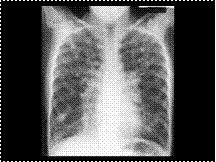
Slide 50
CYSTIC FIBROSIS
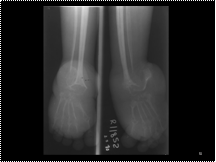
Slide 51
neurofibromatosis
