Spotters B3

Slide 2
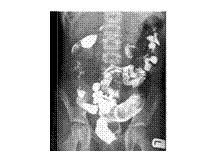
IC KOCHS
Slide 3
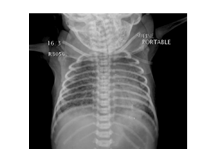
MAS
Slide 4
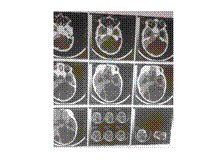
DANDY WAKER
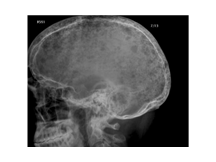
Slide 5
MM
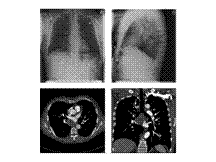
Slide 6
PTE
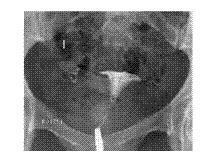
Slide 7
UTERINE SEPTUM
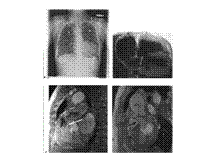
Slide 8
TOF
Slide 9
NOF
Slide 10
GDA PSEUDOANEURYSM
Slide 11
Solid pseudopapillary tumor is also known as a solid-cystic, papillary-cystic, or solid and papillary (epithelial) neoplasm. It is typically benign but may be a low-grade malignancy. These tumors can occur in any part of the pancreas. The incidence is higher in women and these tumors tend to strike relatively young people, with a peak in the second and third decades. Solid pseudopapillary tumors tend to be large (3-18 cm). On gross inspection, there is a thick, fibrous capsule surrounding a solitary, solid to cystic mass (hence the prior terms of solid-cystic tumor). There are typically finger-like projections of tissue of pseudopapillary excresences. Microscopy demonstrates a solid periphery and a pseudopapillary center. As the tumors increase in size, the cystic components—as well as areas of hemorrhage and necrosis—also increase. Ultrasound imaging reveals a solid, hypoechoic, well-circumscribed mass. On CT and MR, the tumor appears heterogeneous (centrally variable) with a thick, enhancing capsule. Papillary projections and hemorrhage may be evident on these modalities.
Serous cystic tumors are also known as microcystic adenoma, serous cystadenoma or cystadenocarcinoma. They are generally benign and occur more often in women than in men (1.5-4:1). Unlike solid pseudopapillary tumors, serous cystic tumors strike older patients, with an average age of 60-70 years. This large (10-13 cm) tumor can occur in any area of the pancreas. On gross examination, it is a spongy, well-cicrumscribed, lobular mass with multiloculated cysts. Microscopy reveals multiple small (<2 cm) cysts containing glycogen-rich fluid, hypervascularity, thin septae and a central stellate calcified scar. On ultrasound, serous cystic tumors are solid lesions with an echogenic central stellate scar. On CT, it is a well-defined, low-attenuation mass that shows marked enhancement; it often has a ""honeycomb"" appearance. On MR, the signal is low dues to the central scar. The mass is hyperintense on T2-weighted images and bright on T1 when areas of hemorrhage are present.
Mucinous cystic tumors are also known as cystadenoma, cystadenocarcinoma, or macrocystic adenoma. They range from benign to frankly malignant and show a strong female predominance (8-9:1). The average age at presentation is 40-60 years. Mucinous cystic tumors occur most often in the tail or body of the pancreas, but can also occur in the pancreatic head. The size is variable, but the cysts contained within the tumor can be large (up to 5 cm). Grossly, the tumor may be either multilocular or unilocular, mimicking a pseudocyst. On microscopy, the tumor is shown to contain cysts >2 cm with a large amount of mucin. The walls are 1-2 mm thick, and the mass is hypovascular. Solid papillary excrescences are visible. On ultrasound, the lesion has internal spetations and papillary projections. Ductal dilatation may be evident. On CT, the mass is hypovascular. The walls and septations enhance with contrast. The appearance on MR is variable and depends upon the material within the cysts (e.g., protein, blood).
Intraductal papillary mucinous tumor (IPMT) is also known as mucin-producing tumor, intraductal mucin hypersecreting neoplasm, mucin hypersecreting tumor, mucinous ductal ectasia or ductectatic mucinous cystic tumor. This is a relatively newly reported entity. It is a soft, villous tumor typically associated with Wirsung’s duct and branches. It is characterized by papillary projections lined by columnar mucin-secreting cells with varying degrees of cellular atypia. The main imaging characteristic is the dilation of the main pancreatic duct, which may or may not be accompanied by a visible cystic lesion
Slide 12
JEJUNAL DIVERTICULI
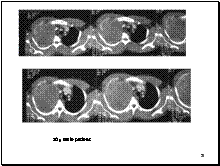
Slide 13
Ewings rib
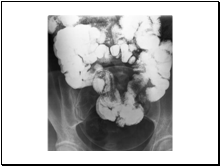
Slide 14
ROUNDWORM
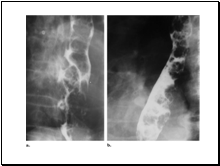
Slide 15
Carcinoma of the Esophagus: Varicoid Pattern1
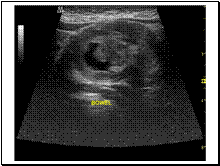
Slide 16
INTUSSECEPTATION
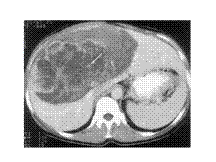
Slide 17
Portal-venous-phase contrast-enhanced CT scan shows a 10-cm-diameter cystic lesion with septa in the right lobe of the liver. Note the calcifications (arrow) within the mass
Slide 18
ARM
Slide 19
HYPOTHYROIDISM
Slide 20
NEUROGENIC TUMOUR
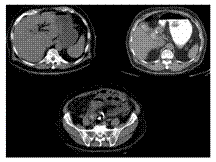
Slide 21
Gallstone ileus
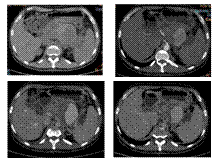
Slide 22
SPLENIC A. PSEUDOANEURYSM
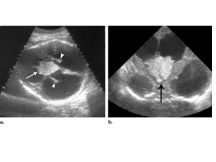
Slide 23
CP PAPPILOMA
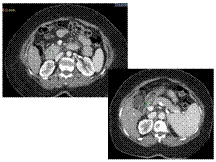
Slide 24
Annular pancreas
Slide 25
MS
Slide 26
Crohn disease
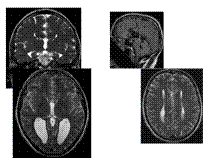
Slide 27
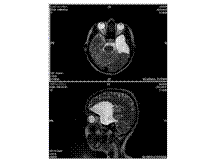
Agenesis of Corpus Callosum
Agenesis of corpus callosum (callosal dysgenesis) accounts for 4% of CNS malformations.
Occurs as an isolated anomaly. Commonly associated with CNS malformations.
A myriad of CNS abnormalities may have callosal dysgenesis as part of their phenotype, including chromosomal abnormalities (trisomy 13), midline abnormalities (Dandy-Walker and Arnold-Chiari malformation), and syndromes associated with mutations in neural adhesion molecules guiding axon growth and migration.
The etiology is due to a failure of axonal formation or migration and may be induced by environmental factors such as fetal alcohol exposure (affecting neuronal cell adhesion molecules), intrauterine infections, and inborn errors of metabolism such as phenylketonuria.
Presentation is usually in childhood. Most children are normal at birth but begin to show progressive signs of subtle cognitive deficits. Prenatal diagnosis is possible using intrauterine ultrasound or MR imaging.
Key Diagnostic Features: Parallel, widely spaced lateral ventricles. Dilated occipital horns (colpocephaly). Radially arranged gyri ""pointing"" to the 3rd ventricle seen on sagittal T1WI, as well as absence of the cingulate gyrus. Coronal sections show characteristic ""trident-shaped"" anterior horns with a ""moose head"" appearance, vertical hippocampi, and ""keyhole"" shaped temporal horns.
DDx: Destruction of the corpus callosum, e.g., Surgery-callosotomy; stretched corpus callosum, e.g., hydrocephalus-thinned corpus callosum but all parts present; and immature corpus callosum
Rx: Treat associated endocrine abnormalities or seizures
Slide 28
Pancreatic #
Slide 29
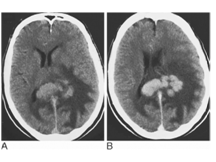
Primary cerebral lymphoma. CT before (A) and after IV contrast medium (B). An irregular mass that is hyperdense to grey matter expands the splenium of the corpus callosum and extends into the left hemisphere. It is surrounded by extensive white matter oedema and enhances avidly with contrast
Slide 30
AAA
Slide 31
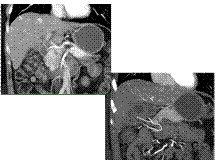
Neuroendocrine Tumor Pancreas With Liver Metastases
Slide 33
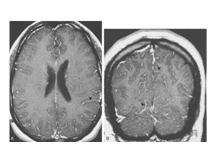
Aseptic meningitis
Slide 34
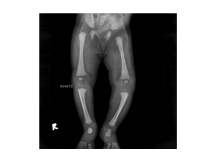
OSTEOPETROSIS
Slide 35
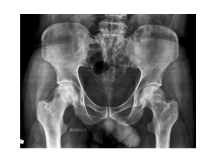
AVN
Slide 36
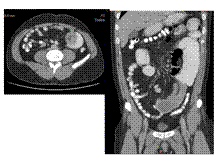
intussception with dupli cyst in jejunum
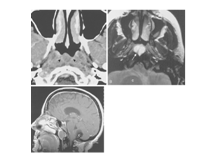
Slide 37
Thornwaldt's cys
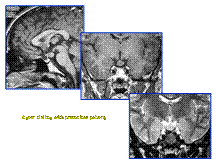
Slide 38
Hamartoma of Tuber Cinerium
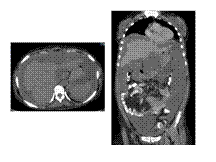
Slide 39
PSEUDOMYXOMA PERITONEA
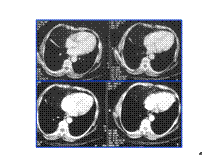
Slide 40
Morgagni Hernia
Slide 41
Arachnoid Cysts